

Este contenido forma parte de la sección Dosis Científicas, en concreto, del especial sobre el grupo hemo. Puedes consultar todas las publicaciones haciendo click aquí.
Patologías relacionadas con la formación del grupo hemo: Porfirias
La síntesis del grupo hemo, como todo proceso en el organismo, pasa por muchas etapas y numerosos compuestos intermediarios.
Además, dadas las importantes funciones que desempeña el hemo, es vital que sea un proceso altamente regulado, ya que defectos en la ruta pueden producir la acumulación de sustancias intermediarias y detener la biosíntesis sin llegar a formar un grupo hemo funcional: estas patologías en la síntesis del grupo hemo son las denominadas porfirias.
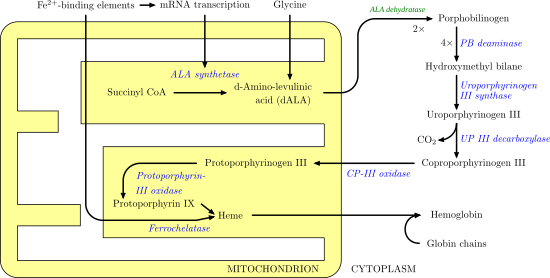
Proceso biosintético de la síntesis del grupo hemo. Se puede ver que tiene lugar en dos compartimentos dentro de la célula (mitocondria y citosol).
Como podemos ver en el diagrama, la síntesis del grupo hemo tiene lugar en muchas etapas y, además, vemos que NO SIEMPRE hay porfirinas (compuestos con 11 dobles enlaces conjugados y por tanto, con capacidad de absorber luz y producir color), sino que se forman porfirinógenos (que son aquellos que no presentan por tanto esa capacidad de conjugación).
Pues bien, tal y como adelantaba en el apartado final de la importancia de los dobles enlaces, defectos en enzimas de esta ruta (las enzimas las vemos marcadas en el esquema en un color azul) van a conllevar la acumulación de productos que, en función de si son porfirinas o no, pueden producir fotosensibilidad al enfermo.
Veamos esto con alguna de estas patologías:
Porfiria eritropoyética congénita (o enfermedad de Günther)
Es una enfermedad que se produce por un defecto en la enzima Uroporfirinógeno III cosintasa. En vez de producirse uroporfirinógeno III se forman dos productos (uroporfirinógeno I y coproporfirinógeno I) que automáticamente se convierten en uroporfirina I y coproporfirina I.
Síntomas
Como he comentado ya, las porfirinas poseen esa capacidad de conjugación de sus dobles enlaces y de absorción de luz. Las personas con defectos en esta enzima, al acumular sustancias de este tipo, van a tener una piel muy sensible a la luz. Esto, tal como podemos leer en la descripción de la PEC en la web de la European Porphyria Network (Red europea de estudios para la Porfiria) «es la causa de que la piel se torne frágil y aparezcan ampollas o úlceras, sobre todo en las zonas más expuestas al sol, tales como cara, dorso de las manos y orejas».
Además, como estas porfirinas se eliminan por la orina, esta adquiere un color rojizo (síntoma muy claro ya desde etapas postnatales).
Otro de los síntomas de la enfermedad de Günther es la anemia hemolítica, producida por la destrucción masiva de glóbulos rojos por el bazo.
Y también pueden presentar retracción de los labios, así como dientes con una coloración rojiza o marrón por la acumulación de estas porfirinas de tipo I.
¿Cómo se transmite generación tras generación esta enfermedad?
La PEC es una enfermedad autonómica recesiva.
- Autosómica porque se localiza en los cromosomas NO sexuales.
- Recesiva porque para que se pueda transmitir, tienen que heredarse dos copias del mismo gen mutado (una del padre, otra de la madre).
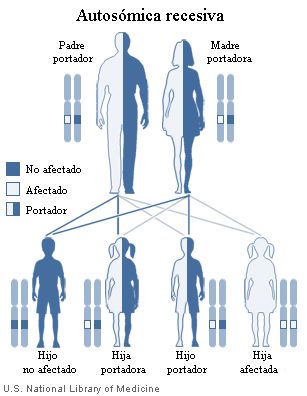
Diagrama de una herencia autosómica recesiva: Podemos ver cómo la hija afectada tiene las 2 copias del gen mutado (representadas con color blanco). Estas copias derivan de ambos progenitores: una del padre y otra de la madre.
Diagnóstico
Se miden los niveles de porfirinas en sangre, orina y heces. A ser posible, se mantendrán las muestras envueltas o tapadas para evitar el contacto con la luz hasta que se realice el análisis.
Puede analizarse también si existe alguna mutación en el gen de la enzima Uroporfirinógeno III cosintasa (gen UROS) y poder verificar si existe o no un defecto en esta enzima.
Porfiria intermitente aguda
Veamos ahora otra de estas porfirias. En este caso, la porfiria intermitente aguda es consecuencia de una carencia en la enzima porfobilinógeno desaminasa (también conocida como hidroximetilbilano sintasa) que produce una acumulación de delta aminolevulinato y porfobilinógeno, inicialmente en el hígado y luego ya, en orina.
Observamos por tanto una diferencia fundamental con respecto al caso anterior, ya que en esta patología NO se acumulan porfirinas, sino porfirinógenos.
Síntomas
En este caso, como dichos compuestos NO presentan capacidad de conjugación en sus dobles enlaces, NO produce fotosensibilidad: esta es una diferencia clave con la PEC.
Además de no afectar a la piel, otro de sus principales síntomas es el de un dolor abdominal muy fuerte que, puede conllevar diarreas, distensión y náuseas en los casos más severos.
Es importante recalcar que los ataques son bastante difíciles de detectar ya que suelen ser muy poco frecuentes.
¿Cómo se transmite generación tras generación esta enfermedad?
La Porfiria Intermitente Aguda es una enfermedad autonómica dominante.
- Autosómica porque se localiza en los cromosomas NO sexuales.
- Dominante porque se puede transmitir con la herencia de solamente 1 de las copias del mismo gen mutado (bien sea del padre o de la madre).
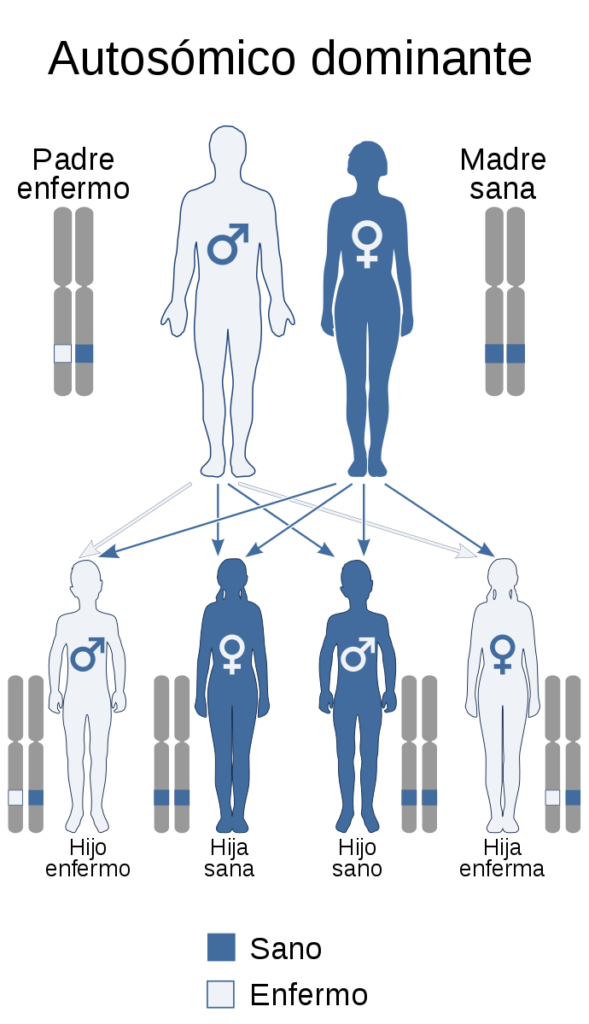
Diagrama donde se explica la herencia autosómica dominante: con que se haya heredado una copia mutada (en el ejemplo, de color blanco), es suficiente para que se transmita la enfermedad a la descendencia. Esa copia puede heredar tanto del padre como de la madre.
Diagnóstico
Antes de nada conviene destacar que «no todas las personas que heredan una mutación genética de estas porfirias desarrollarán un ataque agudo».
Además, parece que las investigaciones al respecto apuntan a que son necesarios ciertos factores externos para que se desencadene un ataque de este calibre (tales como el alcohol, consumo masivo de drogas…).
En cualquier caso, de nuevo, tal y como habíamos comentado con la PEC, se realizan análisis de orina, heces y sangre para medir los niveles de estos compuestos que se acumulan.
Un punto importante que me gustaría recordar es que en la medicina clínica no hay afirmaciones 100% concluyentes, por lo que no es recomendable que se «culpe» a la Porfiria de todos los síntomas (ya que puede haber otras enfermedades, igualmente perjudiciales, que pueden compartirlos (véase algunos tipos de apendicitis…)
Resumen
En conclusión, espero haber podido explicar de una forma más o menos entendible algunos aspectos básicos acerca de las patologías del grupo hemo.
Como siempre decimos, en caso de que tengáis algún síntoma de los que hemos hablado (color en la orina extraño, en las heces…), ya sabéis, no dudéis ni un minuto en consultar con vuestro médico o médica.
¡Son profesionales, recuérdalo!
P.S. Y como siempre, si ha quedado alguna duda, estoy a vuestra disposición para responderlas tanto por correo electrónico como por los comentarios bajo el artículo.
Bibliografía
Diccionario médico-biológico, histórico y etimológico (Dicciomed). Universidad de Salamanca. Ediciones Universidad de Salamanca.
Principios de Bioquímica. Lehninger. David L. Nelson, Michael M. Cox. Ediciones Omega. 7ª edición. 2019
Bioquímica. Conceptos esenciales. Elena Feduchi, Carlos Romero, Esther Yáñez, Isabel Blasco, Carlota García-Hoz. Ed. Médica Panamericana. 2ª edición. 2015






Comentarios